Directory Structure
When using the PEARG clusters or the Melbourne Bioinformatics clusters, your files and data should be organised according to the convention described here.
Each project should have its own directory located inside your home directory or a shared location. Your project directory name should be meaningful.
Any shared data such as reference genomes and indices should be stored outside your project directory to avoid unnecessary duplication of data.
At absolute minimum, please check:
- is your project name sensible?
- do you have a README file (or equivalent) in the parent directory of your project directory that contains meaningful information?
- do you have a data directory (containing your raw data) and a results directory (containing processed data) with files organised in a logical manner?
If you’ve answered no to any of these questions, you may get a stern email in the future.
The motivation behind having a strict structure that all members adhere to for project organisation is transparency. Anyone from the lab should be able to look in your project directory and clearly understand what was run and reproduce your analysis. Most of the time, the person trying to decode what analyses were performed and why will be future you. Cooperate with your future self by leaving verbose notes in README files!
While the sysadmin won’t be authoritarian about the precise directory structure, any flagrant disregard for the guidelines (such as dumping all your processing files in the top-level results directory) will be met with consequences.
Filesystem overview
Click on the image to view a larger version of the recommended directory structure or scroll down for another example in text.
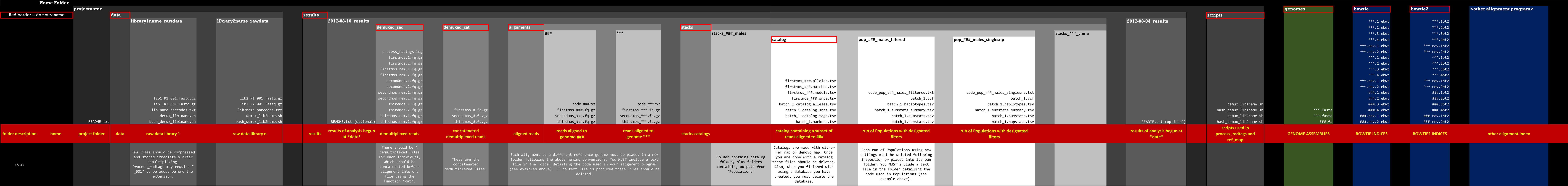
Creating a project directory using the template
# TODO: Jess will create a project skeleton in the future
You should rename the directories starting with rename_* into something
sensible.
Example directory structure
In this example, our project name is called project_name and is a RAD-seq
experiment that was processed using Stacks.
project_name/
├── data/
├── results/
├── scripts/
├── software/
└── README
The directory has four subdirectories: data, results, scripts, and
software, and one README file. The README file should be a plain-text
file containing basic project information (e.g. what the project is about,
what type of data was sequenced).
Data directory
The data directory should contain the raw data received from sequencing.
Each library should have it’s own directory containing sequencing files and
a text file containing barcodes corresponding to samples. This file is needed
for Stacks process_radtags.
project_name/
├── data/
│ ├── library_1_raw_data/
│ │ ├── seqA_R1_001.fastq.gz
│ │ ├── seqA_R2_001.fastq.gz
│ │ └── library_1_barcodes.txt
│ ├── library_2_raw_data/
│ │ ├── seqB_R1_001.fastq.gz
│ │ ├── seqB_R2_001.fastq.gz
│ │ └── library_2_barcodes.txt
├── results/
├── scripts/
├── software/
└── README
Make files read-only (optional)
The files in your data directory should never be edited.
If you are familiar with UNIX file permissions, you can remove write
permissions with the chmod command. For example, the
following command removes write permission for all users:
chmod a-w seqA_R1_001.fastq.gz
You can check file permissions with ls -l where the first column represents
whether read/write/execute access is avaiable.
$ ls -l
-r--r--r-- 1 jess jess 142870 Aug 8 14:30 seqA_R1_001.fastq.gz
-r--r--r-- 1 jess jess 177552 Aug 8 14:30 seqA_R2_001.fastq.gz
Results directory
The results directory should have one directory for each time you generate
a set of results. Using subdirectories inside the main results directory is
recommended because often experiments are re-run in the future (e.g. updated
software versions, more sequencing data, reanalysis before publication).
I recommend naming the directory with a date in YYYY-MM-DD format at the
beginning of the name so the directories are sorted chronologically.
project_name/
├── data/
├── results/
│ ├── 2017-08-01_results/
│ │ ├── ...
│ │ ├── ...
│ │ └── ...
│ ├── 2017-11-01_extra_samples/
│ │ ├── ...
│ │ ├── ...
│ │ └── ...
│ ├── 2018-05-01_reanalysis/
│ │ ├── ...
│ │ ├── ...
│ │ └── ...
├── scripts/
├── software/
└── README
Inside each result subdirectory, there should be multiple directories
containing output from steps in your workflow. In this example, the directories
inside 2017-08-01_results are: demuxed_seq, demuxed_cat, alignments,
stacks, and qc. Your directory names may look different depending on what
type of analysis you’re performing. The contents of each directory is described
below.
Sequencing data
results/
├── 2017-08-01_results/
│ ├── demuxed_seq/
│ │ ├── mozzie-1.1.fq
│ │ ├── mozzie-1.2.fq
│ │ ├── mozzie-1.rem.1.fq
│ │ ├── mozzie-1.rem.2.fq
│ │ ├── mozzie-2.1.fq
│ │ ├── mozzie-2.2.fq
│ │ └── ...
│ ├── demuxed_cat/
│ │ ├── mozzie-1.fq
│ │ ├── mozzie-2.fq
│ │ ├── mozzie-3.fq
│ │ └── ...
│ └── ...
The demuxed_seq directory contains demuxed sequencing data processed by
process_ragtags. Stacks should output four files for each sample listed in
the barcode file. In this example, mozzie-1.1.fq and mozzie-1.2.fq contain
the set forward and reverse reads for the mozzie-1 sample. The
mozzie-1.rem.1.fq and mozzie-1.rem.2.fq files contain the remaining reads
that are unpaired due to their mate being discarded.
If you’re working with ddRADseq data, Stacks recommends concatenating the four
files together. Here, demuxed_cat contains the concatenated files.
Alignment data
results/
├── 2017-08-01_results/
│ ├── demuxed_seq/
│ ├── demuxed_cat/
│ ├── alignments/
│ │ ├── AaegL2/
│ │ │ ├── mozzie-1.AaegL2.sorted.bam
│ │ │ ├── mozzie-1.AaegL2.sorted.bam.bai
│ │ │ ├── mozzie-2.AaegL2.sorted.bam
│ │ │ ├── mozzie-2.AaegL2.sorted.bam.bai
│ │ │ ├── ...
│ │ │ └── AagL2_alignment_code.txt
│ │ ├── AaegL3/
│ │ │ ├── mozzie-1.AaegL3.sorted.bam
│ │ │ ├── mozzie-1.AaegL3.sorted.bam.bai
│ │ │ ├── mozzie-2.AaegL3.sorted.bam
│ │ │ ├── mozzie-2.AaegL3.sorted.bam.bai
│ │ │ ├── ...
│ │ │ └── AagL3_alignment_code.txt
│ └── ...
Alignments should be stored in the alignments directory with a separate
directory for each reference genome aligned against. Alignments should be stored
as bam files with the .bam suffix and bam index files, if provided, should end
with .bai. If alignements are sorted, it’s recommended to include sorted in
the filename. Including the reference genome name in the filename is also
helpful.
A plain-text file with what commands were run should also be included in the
directory (e.g. AagL2_alignment_code.txt) or in the scripts directory.
Stacks data
results/
├── 2017-08-01_results/
│ ├── demuxed_seq/
│ ├── demuxed_cat/
│ ├── alignments/
│ ├── stacks/
│ │ ├── stacks_AaegL2_females/
│ │ │ ├── catalog/
│ │ │ │ ├── mozzie-1.AaegL2.alleles.tsv
│ │ │ │ ├── mozzie-1.AaegL2.matches.tsv
│ │ │ │ ├── mozzie-1.AaegL2.models.tsv
│ │ │ │ ├── mozzie-1.AaegL2.snps.tsv
│ │ │ │ ├── mozzie-2.AaegL2.alleles.tsv
│ │ │ │ ├── ...
│ │ │ │ ├── batch_1.catalog.alleles.tsv
│ │ │ │ ├── batch_1.catalog.snps.tsv
│ │ │ │ ├── batch_1.catalog.tags.tsv
│ │ │ │ └── batch_1.markers.tsv
│ │ │ ├── population_females_filtered/
│ │ │ │ ├── batch_1.vcf
│ │ │ │ ├── batch_1.haplotypes.tsv
│ │ │ │ ├── batch_1.sumstats_summary.tsv
│ │ │ │ ├── batch_1.sumstats.tsv
│ │ │ │ ├── batch_1.hapstats.tsv
│ │ │ │ └── code_females_filtered.txt
│ │ │ ├── population_females_singlesnp/
│ │ │ │ ├── batch_1.vcf
│ │ │ │ ├── batch_1.haplotypes.tsv
│ │ │ │ ├── batch_1.sumstats_summary.tsv
│ │ │ │ ├── batch_1.sumstats.tsv
│ │ │ │ ├── batch_1.hapstats.tsv
│ │ │ │ └── code_females_singlesnp.txt
│ │ ├── stacks_AaegL2_males/
│ │ │ └── ...
│ │ └── ...
│ └── ...
Each run of Stacks to get a catalogue should have its own separate directory
in the stacks directory. The output files from ref_map or denovo_map
stored in its own directory.
Each time you run Stacks populations with designated filters, you should
store the files in a separate directory. You should also include a file
containing the code that was used to produce the output.
QC data
results/
├── 2017-08-01_results/
│ ├── demuxed_seq/
│ ├── demuxed_cat/
│ ├── alignments/
│ ├── stacks/
│ ├── qc/
│ │ ├── fastqc/
│ │ │ ├── ...
│ │ │ ├── ...
│ │ │ └── ...
│ │ ├── flagstat/
│ │ │ ├── ...
│ │ │ ├── ...
│ │ │ └── ...
│ │ └── ...
│ └── ...
The qc directory should contain the output of programs run for quality
control purposes (e.g. fastQC, samtools flagstat).
Scripts directory
It’s up to you if you want to store your scripts inside the scripts directory
or with the output files that were generated. Just make sure you document
all the code that was run somewhere sensible.
Software directory
If you have any additional software you compiled specifically for your project, you can store them here.
README files
README files are plain-text files where you should write descriptions of what the directory contains, what analysis was done, why certain parameters were chosen, what results were found, etc. Place a README file in any directory you feel could use one. Documenting your work clearly is good practice and often pays dividends in the future.